到了 1970 年代末,已經出現兩大類人類癌症病因的證據,但這兩者之間看來並無關聯。病毒致癌基因和原致癌基因漂亮地解釋病毒會引發癌症,但這種癌症只限於動物;某些人類癌症會出現特殊的染色體變異,其一致性很具說服力,但只限於幾種癌症,而且其中涉及的基因並不明朗。那麼,這兩者癌症病因之間有關連嗎?
有的,這個關聯指出,癌症是調節失常所造成的疾病。
調節失常而致癌:失控的正向基因

在最早發現的那些病毒致癌基因和細胞原致癌基因中,在 src 之後找到的是小鼠的 v-abl 基因,這個基因來自於艾貝爾遜白血病病毒(Abelson leukemia virus),它在細胞中相似的基因是 c-abl 基因。
c-abl 和其他 c-src 等基因一樣,也存在於人類的基因組中。然而,後來科學家發現,c-abl 基因位在第九號染色體上,這個染色體也是羅利指出在慢性骨髓性白血症癌細胞中發生轉位的染色體,於是他們猜想:這有關連嗎?那些慢性骨髓性白血症患者癌細胞中第九號染色體斷裂的部位,靠近 c-abl 基因嗎?
這是風險很大的推論。染色體很大,每個平均約有一千個基因,而 c-abl 基因可能存在於任何位置。一個由荷蘭與英國科學家組成的團隊研究了賓州染色體(第二十二號染色體),赫然發現其中有本來在第九號染色體上的 c-abl 基因——這個基因轉位到第二十二號染色體上了(下圖左)。
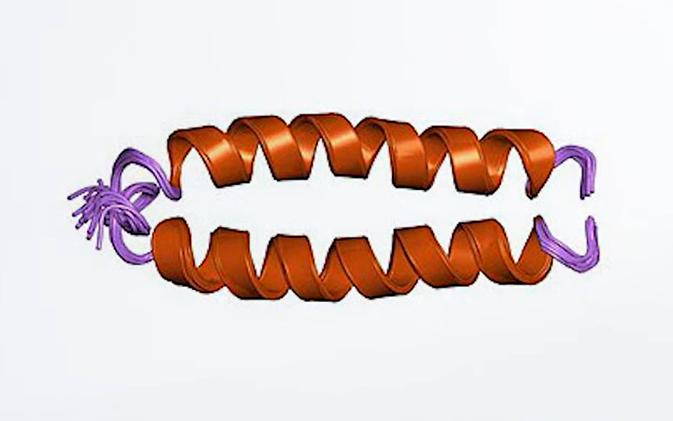
這項發現令人振奮,因為 c-abl 基因和人類癌症很可能有直接關連。研究者找出了第二十二號染色體中和 c-abl 基因相鄰的部位,好知道在慢性骨髓性白血症患者癌細胞中,c-abl 基因到底發生了什麼事。結果發現了值得注意的現象:在十七位病人中,c-abl 基因都轉移到第二十二號染色體上相同的位置。所以不只第九號染色體的片段轉位到第二十二號染色體上很重要,轉位到相同位置上更是重要。這樣的結果指出,c-abl 基因在第二十二號染色體上的位置才是重點。進一步的檢查還發現,c-abl 基因與另一個基因 bcr(意思是「斷裂點簇集區」,”breakpoint cluster region”)連接在一起。這兩個融合在一起的基因會製造一種異常的蛋白質,有著 c-abl 蛋白質的前端和 bcr 蛋白質的後端(下圖右)。
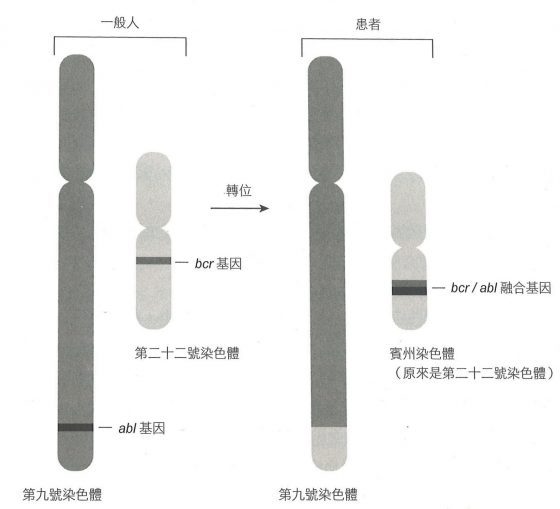
總之,這樣的融合使得正常的原致癌基因變成了致死的致癌基因。研究人員比較正常的 c-abl 蛋白質和 bcr/abl 融合蛋白質兩者的活性之後,發現了箇中道理。c-abl 蛋白質屬於酪胺酸激酶(tyrosine kinases)這類酵素,其功用是把磷酸連接到蛋白質上。在蛋白質上增添或是移除磷酸,是另一種常見的蛋白質活性調節方式,讓蛋白質在活性狀態和無活性狀態之間變化。許多激酶屬於化學傳遞系統的一環,這個系統能把來自細胞外的訊息傳遞到細胞內部,好讓細胞複製、分化或是死亡。在細胞中,c-abl 酪胺酸激酶的活性通常很低,但是融合後的突變蛋白質,就像是莫納德和賈哥布所研究的「持續」突變一樣,總是處於「啟動」的狀態。
所以白血病是一種調節失常造成的疾病。在慢性骨髓性白血症中,原本受到控制的白血球複製功能,會因為 bcr/abl 融合蛋白而失控。這種活性超高的蛋白質會干擾細胞中多條訊息傳遞系統,因此細胞分裂的訊息會一直處於「開啟」狀態,就像是一直踩著油門的汽車。後來科學家發現,其他幾十種致癌基因中發生的突變,都是經由這種普遍的效應,與許許多多其他的癌症扯上關係。這意味癌症通常是調節失常所造成的疾病。
發現致癌基因和它們的運作模式,對於瞭解癌症而言向前邁進了一大步,不過致癌基因只占癌症遺傳故事的一半而已。我們在這本書中已經討論過調節的邏輯,因此你可能會猜到另一半故事的內容。持續加油的油門當然不會是車子失控的唯一原因,那麼另一個機制是什麼呢?(提示:想想負向循環與負負得正的調節邏輯。)
如果你的腳沒有踩煞車,或是煞車線斷了,效果也是一樣。研究人員的確發現,在癌症出現的過程中,失去了遺傳「煞車」,是件稀鬆平常的事。
煞不住的癌症進程:缺席的抑癌基因
第一個遺傳「煞車」,是在一種罕見的眼睛癌症——視網膜胚細胞瘤(retinoblastoma)-—中發現的。這種癌症通常出現在幼兒身上,有時是家族遺傳的。解開視網膜胚細胞瘤遺傳奧秘的重要線索,來自於有些病患的兩個第十三號染色體都失去了一個部分。這表示某些基因的兩個拷貝都沒有了,對視網膜胚細胞瘤的形成而言非常關鍵。這個狀況與致癌基因只要有一個拷貝發生改變(例如 bcr/abl),就足以成為癌症形成的關鍵事件,兩者恰恰相反。
若使用遺傳學的術語,我們會說致癌基因的突變是顯性的,因為即使正常的原致癌基因完好無缺,致癌基因還是會造成影響。相反地,視網膜胚細胞瘤突變是隱性的,因為要兩個基因拷貝都改變,才會使疾病成形。看來那個失去的基因的正常功能,是阻止或是壓抑癌症的形成,因此我們把這種基因稱為「癌症抑制基因」。
科學家集中研究視網膜胚細胞瘤患者失去的 DNA,結果找到了視網膜胚細胞瘤基因(稱為 Rb)。Rb 基因的功能當然不是引發癌症,因為是 Rb 基因消失或變了樣之後,癌症才會出現的。之後研究了 Rb 蛋白質,發現它的功能是控制細胞循環的一個重要部分。細胞要複製時,會先複製 DNA,然後才分裂成兩個;這個過程受到嚴密的調控,而且分成多個階段進行。而 Rb 蛋白質作用在細胞循環早期階段的一個關卡上,能阻止 DNA 的複製。然而,當兩個 Rb 基因都沒有了的時候,細胞就能不受控制地持續複製。
Rb 不是唯一能抑制癌症的基因,現在科學家已找出約七十個這樣的基因。Rb 也不只和視網膜胚細胞瘤有關,其他癌症中也見到了Rb 突變,例如骨肉瘤(osteosarcoma)和肺癌。
讓 Rb 失去活性的方法不只突變而已,當激酶把磷酸根加到 Rb 蛋白質上,也可以調節 Rb 蛋白質的活性-—上面的磷酸越少,活性越高;磷酸越多,活性就越低。許多致癌基因(包括 bcr/abl)直接或間接的效應,是讓 Rb 蛋白上的磷酸增加而使得活性受到抑制,這樣細胞就會持續複製。事實上,幾乎在所有人類的癌症中,Rb 蛋白的活性都受到某種程度的抑制。
這裡又出現了我們之前見過的那種負向調節和負負得正調節邏輯。一般來說,Rb 蛋白能抑制細胞增殖;細胞增殖通常需要抑制這種抑制蛋白,才能繼續進行。不過 Rb 蛋白如果沒有活化(左)或是缺失了(右),細胞就會持續增殖:
幾十年前,莫納德與賈哥布就推測,癌症是因為細胞複製的抑制子失去活性所造成的,Rb 蛋白的角色完全符合他們的推測(見本書第三章)。
知道了某些基因的突變會打破細胞生長的調節機制,那麼下一個重大的挑戰,就是想辦法把癌症細胞的「剎車」裝回去。
從實驗室走向臨床製藥
幾十年來,癌症療法通常不是用手術切除腫瘤,就是用放射線和混合各種藥物來殺死分裂中的細胞。後者是盲目攻擊,無法特別針對癌細胞,導致療效差異性高,因此使用受到限制,而各種副作用會讓人衰弱,甚至引發危險。因此,癌症研究一直致力於設計出可針對病人特定癌症治療,同時更有效、更安全的療法。現在這種希望成真了;這類藥物最先上市的是基利克(Gleevec),作用的目標就是當年羅利在餐桌上找到的突變。
基利克就像其他類別的疾病最先出現的藥物那樣,差點夭折在研發的半路上。事實上,基利克的故事和第一個史達汀藥物的研發歷史相似到可怕。這次也是由於一位醫生他瞭解病人的需求,努力不懈地鼓勵藥物研發,才出現這個改變醫療歷史的偉大臨床成就。
bcr/abl 的基因轉位,產生了一個活性超強的激酶,使得 Rb 蛋白的抑制功能無法活化,導致細胞分裂不受控制。我們需要的是能搞定慢性骨髓性白血症負負得正邏輯的藥物——能抑制 bcr/abl 融合蛋白的作用,讓這變節的酵素無法造成傷害。
萊登(Nick Lydon)與馬特(Alex Matter)這兩位在瑞士巴賽爾的汽巴—嘉基製藥公司(Ciba-Geigy)任職的科學家,知道許多致癌基因的產物是變異的激酶,那麼這些酵素的抑制劑應該能阻止癌細胞生長。他們不像遠藤那樣在大自然中尋找,或是依循製藥工業傳統的嘗試錯誤方式,而是使用稱為「理性設計」(rational design)的策略,設計出能夠緊密嵌入激酶活性部位並抑制活性的分子。如此一來,一般的「鑰匙」就沒辦法插入「鎖孔」中了。經過多年的化學合成與測試,他們得到了幾種有潛能的分子,其中一種分子能夠抑制正常的 c-abl 激酶。
萊登把這些化合物提供給一位認識的醫生,好測試其中是否有能對付慢性骨髓性白血症癌細胞的分子。這位在美國波特蘭奧勒岡健康與科學大學(Oregon Health & Science University)任職的醫生杜魯克(Brian Druker),對可能抑制 bcr/abl 激酶活性的化合物深感興趣;更重要的是,他可以取得慢性骨髓性白血症患者的細胞。杜魯克發現,萊登給他的某種化合物在非常低的濃度下,能殺死這些細胞,但是正常的細胞不會死亡。

就在萊登、馬特和杜魯克為此結果興奮不已時,製藥公司認為專門治療慢性骨髓性白血症的藥物沒有市場,這讓他們花了一年多的時間,才說服公司進一步進行動物實驗。首次在狗身上進行毒理測試的結果,讓人擔心這種藥物若在人類身上以靜脈注射的方式施用,可能並不安全。之後過沒多久,汽巴—嘉基製藥公司和山德世製藥公司(Sandoz)合併,成立了新的公司諾華(Novartis)。公司合併之後,這個藥物的發展一蹶不振,萊登也辭職了。
癌症標靶療法問世
後來諾華的科學家用口服方式在狗身上持續進行試驗,但結果還是不行。有位毒物學家告訴馬特:「除非我死了,否則這個化合物不能給人使用。」
杜魯克沒有被結果嚇到,他的病人預後狀況非常糟糕:在診斷出罹患這個疾病之後,有 25~50% 會在一年內死亡,他能做的最多只是用現有療法稍微延長病人的壽命。杜魯克認為,藥物的毒性可以經由監測病人並且改變劑量來控制,因此他催促馬特「再給這個藥物一個機會」,馬特也持續對公司管理階層勸說這種藥物的需求。最後諾華的新任執行長華塞拉(Daniel Vasella)支持這個藥物進行人體試驗,這項研究在 1998 年 6 月展開,距離杜魯克在實驗室中用慢性骨髓性白血症癌細胞測試這個藥物,已經相隔五年。
杜魯克和其他兩位醫生開始在少數慢性骨髓性白血症患者身上使用這種藥物,並且逐漸增加藥物劑量,同時觀察患者的病況以及可能出現的副作用。這個藥物如果有效,可以從白血球數量的減少程度看出來。正常人的白血球數量是每微升(microliter, μL)血液中有四千到一萬個,但是慢性骨髓性白血症患者會飆升到十萬到五十萬個。在藥物劑量低的時候,他們沒有觀察到療效,但在增高劑量之後,卻發現有些病人的白血球數量下降到正常範圍。用顯微鏡觀察病患的血液,可以看到帶有賓州染色體的細胞所占的比例減少了。這表示,這個藥物能夠成功殺死目標。
諾華把所有資源都投入在發展這個藥物上,實驗規模擴大,劑量增加,同時追蹤病患好幾個月。使用高劑量的病人有 97% 在六周後白血球數量恢復正常,四分之三的病人體內含有賓州染色體的細胞消失了。這不只是好結果,而是非常了不起的結果,在癌症化療上史無前例。美國食品與藥物管理局優先審查這個藥物,不到三個月就核准上市了,時間是 2001 年 5 月。
有了基利克,慢性骨髓性白血症預後的狀況大幅改善,長期存活率(超過八年)躍升到 90%,而沒有使用這個藥物時只有 45%。和諾華公司預期的相反,這個藥物成為該公司的暢銷藥物,十年來銷售額達到二百八十億美元。2012 年,萊登、杜魯克和羅利因為對慢性骨髓性白血症的研究和治療,獲得著名的日本國際獎(Japan Prize)。

本文摘自《生命的法則:在賽倫蓋蒂草原,看見大自然如何運作》,八旗文化出版。